4.1 Ecological Selectivity and the Expanding Landscape of Microbial Recovery
The synthesis of evidence across the reviewed studies revealed that microbial recovery in both environmental microbiology and clinical metagenomics is governed less by simple detection capability and more by the ability to selectively enrich biologically meaningful signals from overwhelmingly complex backgrounds. Interestingly, this challenge appeared remarkably consistent across vastly different ecosystems. Whether researchers were isolating rare Streptomyces strains from hyper-arid soils or attempting to identify low-abundance pathogens in host-dominated clinical samples, the same underlying obstacle repeatedly emerged: valuable microbial signatures were often concealed beneath dominant non-target genetic material.
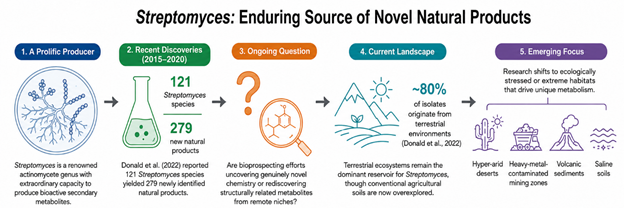
The cultivation strategies summarized in Table 1 demonstrated how environmental microbiologists increasingly rely on ecological filtering to recover rare actinomycetes from highly competitive microbial communities. Pretreatment conditions involving salinity adjustment, selective antibiotics, pH manipulation, and oxidative stress appeared particularly effective for enriching resilient Streptomyces species adapted to extreme ecological niches. As shown in Table 1, isolates such as S. boncukensis, S. arcticus, and S. canalis were recovered using combinations of cycloheximide, potassium dichromate, or elevated NaCl concentrations, suggesting that environmental stress itself may function as a selective gateway for discovering metabolically specialized microorganisms (Donald et al., 2022). In many respects, these approaches resemble a form of controlled ecological pressure rather than straightforward laboratory cultivation.
The findings further suggested that selective enrichment strategies may unintentionally shape the diversity of organisms ultimately recovered. Dry heat treatment, antifungal suppression, and nutrient limitation effectively reduced fast-growing competitors, yet there remains some uncertainty regarding how many biologically valuable organisms are simultaneously excluded through these aggressive filtering processes. Stewart (2012) and Vartoukian et al. (2010) previously emphasized that most environmental microorganisms remain unculturable under standard laboratory conditions, and the present synthesis reinforces that concern. Selective isolation undoubtedly improves recovery efficiency for resistant taxa, but it may also narrow the observable microbial landscape in ways that remain poorly understood.
The comparative data presented in Table 1 therefore highlight an important conceptual parallel with clinical host DNA depletion workflows. Differential lysis and nuclease digestion strategies used in metagenomic sequencing operate according to nearly identical principles: fragile host cells are selectively disrupted while structurally resilient microbial cells remain intact long enough for downstream sequencing. Although developed independently, both environmental cultivation and clinical depletion strategies appear fundamentally rooted in the same biological logic—preferential survival through selective pressure.
4.2 Secondary Metabolite Discovery and the Hidden Biosynthetic Reservoir
The studies summarized in Table 2 collectively revealed that environmental and host-associated microorganisms continue to represent extraordinarily rich reservoirs of unexplored biochemical diversity. Yet the findings also suggested that conventional discovery pipelines may capture only a relatively small fraction of total biosynthetic potential. Many of the compounds identified through metagenomic and sequence-guided approaches originated from organisms that were previously inaccessible through standard cultivation methods.
Several metabolites listed in Table 2 illustrated the remarkable pharmacological diversity associated with environmental microbiomes. Teixobactin, isolated from Eleftheria terrae using iChip cultivation technology, demonstrated potent activity against Staphylococcus aureus without detectable resistance development during initial studies (Mohimani et al., 2018). Similarly, malacidin A and lassomycin highlighted the growing importance of metagenomics for recovering antimicrobial compounds from uncultured bacteria. These findings reinforce the broader realization that microbial “dark matter” may contain substantial untapped therapeutic potential.
Host-associated microbiomes appeared particularly significant in this regard. The tunicate-associated Streptomyces strains summarized in Table 2 produced metabolites such as platensimycin B with activity against methicillin-resistant pathogens including MRSA and Enterococcus faecium (Utermann et al., 2020). Likewise, marine sponge-associated bacteria described by Almeida et al. (2023) exhibited broad biosynthetic versatility and antimicrobial potential. These observations suggest that host-associated ecosystems may promote unusually specialized metabolic adaptation driven by intense microbial competition and ecological interdependence.
At the same time, the findings revealed a growing methodological transition from phenotype-dependent discovery toward sequence-guided identification strategies. Traditional culture-based screening remains valuable, yet repeated rediscovery of previously characterized metabolites continues to limit its efficiency (Genilloud et al., 2011). In contrast, metagenomic sequencing and biosynthetic gene cluster prediction increasingly allow researchers to identify theoretically valuable pathways before metabolite expression is experimentally confirmed.
This transition became especially evident in compounds such as landepoxin A, where computational sequence analysis identified biosynthetic signatures prior to functional validation (Mohimani et al., 2018). Such findings suggest that microbial genomes may encode extensive “silent” chemistry that remains inaccessible under standard laboratory conditions but detectable through genomic mining. Consequently, the distinction between discovery and prediction is becoming progressively blurred within modern metagenomic research.
The results summarized in Table 2 therefore indicate that microbial secondary metabolism extends far beyond what can be observed experimentally through conventional cultivation alone. Increasingly, metagenomics appears to function not merely as a sequencing tool but as a predictive framework for identifying hidden biochemical potential embedded within complex microbial communities.
4.3 Expansion of Bioinformatic Genome Mining and Computational Dereplication
The reviewed evidence further demonstrated that bioinformatics has evolved from a supportive analytical tool into one of the central driving forces behind contemporary metagenomic interpretation. As sequencing
Table 2. Natural Product Discovery and Biological Activities from Environmental and Host-Associated Microorganisms. This table presents representative secondary metabolites identified through culture-dependent, metagenomic, and sequence-guided discovery pipelines across terrestrial, marine, and host-associated microbial systems. The data emphasize the diversity of bioactive compounds produced by environmental microorganisms and demonstrate how approaches such as iChip cultivation, metagenomics, GC-MS, and UPLC-MS/MS have expanded access to previously hidden biosynthetic potential.
|
Compound Name
|
Chemical Class
|
Producing Organism
|
Habitat
|
Targeted Pathogen
|
Bioactivity
|
Detection Method
|
Reference
|
|
Chaxalactin A
|
Macrolactone PK
|
Streptomyces C34
|
Desert soil
|
—
|
Medium-dependent activity
|
Culture-based screening
|
Mohimani et al. (2018)
|
|
Teixobactin
|
Antibiotic
|
Eleftheria terrae
|
Soil
|
S. aureus
|
No detectable resistance
|
iChip
|
Mohimani et al. (2018)
|
|
Lassomycin
|
Cyclic peptide
|
Lentzea sp.
|
Soil
|
M. tuberculosis
|
ATP-dependent activity
|
iChip
|
Mohimani et al. (2018)
|
|
Malacidin A
|
Lipopeptide
|
Uncultured bacterium
|
Soil
|
Gram-positive bacteria
|
Antibacterial activity
|
Metagenomics
|
Mohimani et al. (2018)
|
|
Landepoxin A
|
Epoxyketone
|
Metagenomic clone
|
Soil
|
—
|
Proteasome inhibition
|
KS-tag sequencing
|
Mohimani et al. (2018)
|
|
Arimetamycin A
|
Anthracycline
|
Streptomyces albus
|
Soil
|
—
|
Cytotoxic activity
|
Sequence-guided screening
|
Mohimani et al. (2018)
|
|
TP-1161
|
Thiopeptide
|
Nocardiopsis sp.
|
Marine environment
|
Gram-positive bacteria
|
Vancomycin-like activity
|
OSMAC
|
Mohimani et al. (2018)
|
|
2,3-Butane diol
|
Diol
|
SP7, SP9 isolates
|
Saltpan
|
—
|
Antifreeze activity
|
GC-MS
|
Ramganesh et al. (2017)
|
|
Borinic acid
|
Organic acid
|
SP8 isolate
|
Saltpan
|
Acne-associated bacteria
|
Dermatological potential
|
GC-MS
|
Ramganesh et al. (2017)
|
|
Platensimycin B
|
Diterpenoid
|
Streptomyces sp.
|
Tunicate gut
|
MRSA, E. faecium
|
Antibacterial activity
|
UPLC-MS/MS
|
Utermann et al. (2020)
|
Table 3. Bioinformatic Tools and Pipelines for Metagenomic Mining and Biosynthetic Gene Cluster Identification. This table summarizes widely used computational platforms designed for metagenomic dereplication, biosynthetic gene cluster (BGC) prediction, peptide identification, and phylogenetic classification. These tools collectively support sequence-guided discovery by enabling rapid annotation of NRPS, PKS, RiPP, and cryptic biosynthetic pathways from increasingly large and complex metagenomic datasets.
|
Tool Name
|
Input Data
|
Output Type
|
BGC Class
|
Unique Feature
|
Accessibility
|
Major Advantage
|
Reference
|
|
antiSMASH 4.0
|
Genome sequence
|
Prediction/BLAST output
|
NRPS, PKS
|
Cluster boundary identification
|
Web server
|
Comprehensive annotation
|
Mohimani et al. (2018)
|
|
PRISM 3
|
Genome sequence
|
Chemical structure prediction
|
NRPS, PKS
|
Tailoring reaction prediction
|
Open-source
|
Structural identification
|
Mohimani et al. (2018)
|
|
SeMPI
|
Genome sequence
|
Top 10 matches
|
Type I PKS
|
Non-modified PKS analysis
|
Web application
|
Best-match comparison
|
Mohimani et al. (2018)
|
|
Pep2Path
|
Genome sequence
|
Mass-shift analysis
|
NRPS, RiPPs
|
Tag-BGC alignment
|
Open-source
|
Automated peptide linking
|
Mohimani et al. (2018)
|
|
RiPPquest
|
Genome sequence
|
Peptide identification
|
RiPPs
|
MS/MS alignment
|
Open-source
|
Peptidic compound detection
|
Mohimani et al. (2018)
|
|
NRPquest
|
Genome sequence
|
Peptide identification
|
NRPS
|
Mass spectrometry coupling
|
Open-source
|
NRPS characterization
|
Mohimani et al. (2018)
|
|
NaPDos
|
Sequence tags
|
Phylogenetic classification
|
NRPS, PKS
|
Domain-based search
|
Web tool
|
Phylogenetic analysis
|
Mohimani et al. (2018)
|
|
eSNAPD
|
Sequence tags
|
NPST library generation
|
—
|
Diversity survey platform
|
Tool set
|
Large dataset processing
|
Mohimani et al. (2018)
|
|
ClustScan
|
Metagenomic data
|
Annotation output
|
Modular BGCs
|
Homology searching
|
Software package
|
Semi-automated analysis
|
Chen et al. (2019)
|
|
ClusterFinder
|
Metagenomic data
|
Novel cluster prediction
|
Unspecified
|
Hidden BGC detection
|
Algorithm-based
|
Novelty-focused discovery
|
Chen et al. (2019)
|
Table 4. Host-Associated Microbiome Diversity and Metagenomic Functional Potential This table highlights the microbial diversity, biosynthetic potential, and ecological specialization observed within host-associated microbiomes and environmentally complex microbial communities. The compiled studies demonstrate how marine invertebrates, estuarine sediments, geothermal systems, and insect-associated microbiota function as reservoirs of biosynthetic gene clusters linked to antimicrobial, anticancer, cytotoxic, and ecosystem-regulating activities.
|
Sample Source
|
Location
|
Dominant Microbes
|
Genera Count
|
Sequenced Strains/Features
|
Key SM-BGCs
|
Functional Potential
|
Reference
|
|
Ciona intestinalis
|
Baltic/North Sea
|
Gammaproteobacteria
|
33
|
101 isolates
|
Peptides, PKs
|
Anticancer and antimicrobial activity
|
Utermann et al. (2020)
|
|
Marine sponges
|
Taiwan
|
Proteobacteria
|
55
|
70 genomes
|
RiPPs, NRPS, PKS
|
Anti-Candida activity
|
Almeida et al. (2023)
|
|
T. swinhoei
|
Marine environment
|
Uncultured symbionts
|
—
|
Entotheonella spp.
|
Polytheonamides
|
Cytotoxic peptide production
|
Mohimani et al. (2018)
|
|
Didemnum molle
|
Papua New Guinea
|
Cyanobacteria
|
—
|
Prochloron spp.
|
Divamides
|
Anti-HIV activity
|
Mohimani et al. (2018)
|
|
Estuary sediment
|
Pearl River
|
Archaea, Bacteria
|
—
|
ASVs quantified
|
Chitin turnover pathways
|
Environmental adaptation
|
Zvi-Kedem et al. (2022)
|
|
Geothermal waters
|
Croatia
|
Eukaryota
|
—
|
V9 rRNA regions
|
Protist diversity
|
Ecosystem stability
|
|
|
Soil microbiomes
|
Global (five continents)
|
NRPS, PKS producers
|
—
|
Diverse microbiota
|
PKs and NRPs
|
Antimicrobial resistance ecology
|
Chen et al. (2019)
|
|
Saltpan water
|
South Africa
|
Bacteroidetes
|
—
|
SP1–SP22 isolates
|
—
|
Hydrocarbon degradation
|
Ramganesh et al. (2017)
|
|
Camponotus ants
|
China
|
Streptomyces spp.
|
—
|
Ant-associated strains
|
NRPS, RiPPs
|
Symbiotic defense
|
Donald et al. (2022)
|
|
Marine invertebrates
|
Orkney, Scotland
|
Gammaproteobacteria
|
23
|
77 extracts
|
Metabolites <1000 Da
|
Novel metabolomic diversity
|
Macintyre et al. (2014)
|
capacity increased dramatically, the primary bottleneck shifted away from data generation and toward the identification of biologically meaningful information hidden within massive genomic datasets.
The computational platforms summarized in Table 3 collectively illustrated how modern genome-mining tools now enable rapid annotation of biosynthetic gene clusters (BGCs), peptide pathways, and cryptic metabolic signatures. antiSMASH and PRISM emerged as particularly influential platforms because of their ability to identify conserved biosynthetic architectures directly from genome sequences (Mohimani et al., 2018). These tools substantially reduced analytical redundancy by rapidly distinguishing previously characterized pathways from potentially novel biosynthetic systems.
Interestingly, different computational frameworks appeared to prioritize different forms of discovery. antiSMASH emphasized broad-spectrum annotation and comparative genomic mapping, whereas ClusterFinder focused more aggressively on detecting atypical or low-similarity pathways that might otherwise remain overlooked (Chen et al., 2019). While this increased the possibility of false-positive predictions, it also improved the likelihood of identifying genuinely novel biosynthetic chemistry.
The importance of dereplication became especially apparent in host-associated microbiome studies. Utermann et al. (2020) reported annotation efficiencies approaching 71% in tunicate-associated microbial systems, substantially accelerating prioritization of candidate pathways for downstream characterization. Similarly, NaPDos and eSNAPD provided phylogenetic and diversity-based frameworks capable of processing increasingly large environmental sequence datasets.
These findings suggest that bioinformatics is no longer functioning solely as a post-sequencing cleanup mechanism. Rather, computational dereplication now actively shapes how microbial diversity is interpreted, prioritized, and biologically contextualized. The ability to computationally suppress redundant information while enriching low-frequency genomic signals increasingly mirrors the conceptual goals of physical host DNA depletion itself.
4.4 Host-Associated Microbial Ecosystems and Functional Specialization
One of the more striking findings emerging from the reviewed literature involved the ecological complexity of host-associated microbial communities. Marine invertebrates, tunicates, and sponge-associated microbiomes consistently demonstrated unusually high levels of microbial specialization and biosynthetic diversity. These systems appear less like simple microbial colonization events and more like highly integrated ecological networks shaped by long-term coevolutionary pressures.
The microbial ecosystems summarized in Table 4 revealed substantial functional specialization across both environmental and host-associated microbiomes. Sponge-associated bacteria exhibited extensive NRPS, PKS, and RiPP biosynthetic capabilities, while tunicate-associated communities produced metabolites linked to antimicrobial and anticancer activities (Almeida et al., 2023; Utermann et al., 2020). In many cases, microbial symbionts appeared to function as biochemical defense partners that directly contribute to host survival.
Zvi-Kedem et al. (2022) further demonstrated that estuarine microbial systems display pronounced ecological structuring driven by environmental gradients and host interactions. Similarly, geothermal water microbiomes revealed unexpectedly diverse eukaryotic microbial populations capable of maintaining ecological stability under extreme physicochemical conditions.
These findings collectively reinforce the idea that host-derived material should perhaps not be viewed exclusively as undesirable analytical interference. Instead, host-associated environments appear to actively shape microbial adaptation, biosynthetic specialization, and metabolic expression. This perspective may carry important implications for clinical metagenomics, where host DNA depletion strategies have traditionally focused primarily on subtraction rather than ecological interpretation.