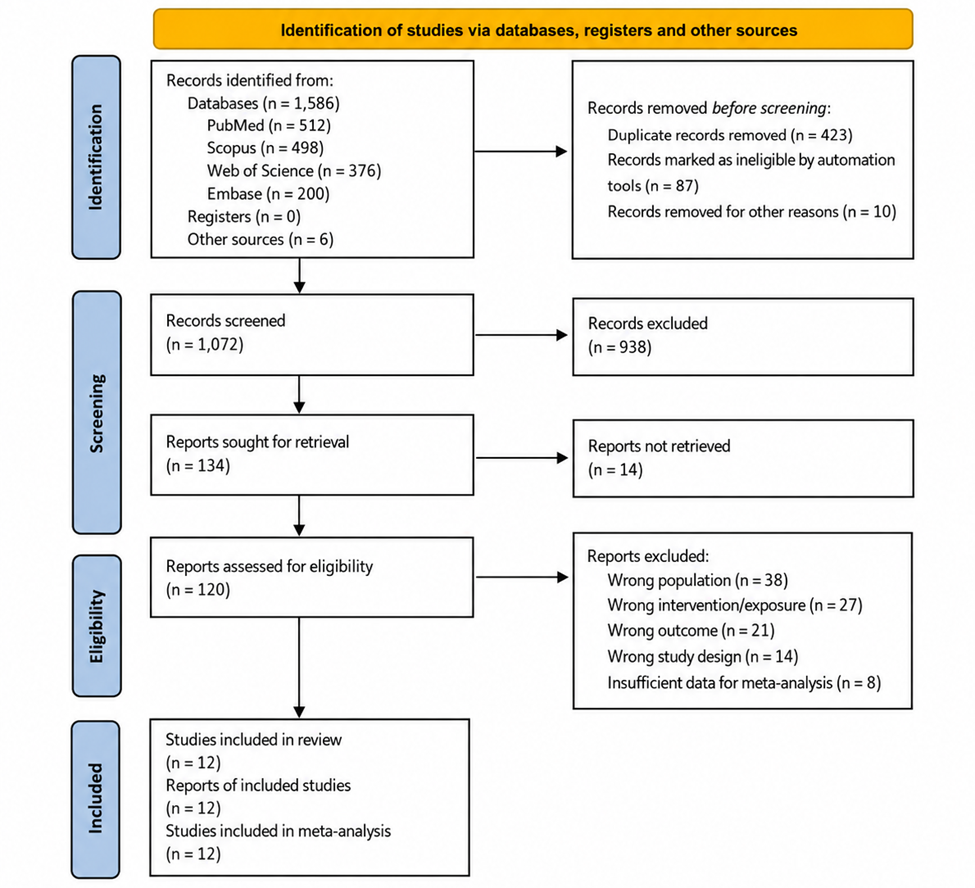
1. Introduction
The exploration of microbial communities, or microbiomes, and their associated pathogens represents a central frontier in contemporary biological sciences. Microorganisms inhabit virtually every environment on Earth, from nutrient-rich soils to the extreme pressures of hadal oceanic zones, and their interactions shape both ecosystem functions and host health. Historically, our understanding of microbial ecology was heavily constrained by culture-dependent techniques, which allowed the study of merely a fraction of microbial life. It is estimated that approximately 99% of prokaryotic species remain uncultivable under standard laboratory conditions, creating a vast “microbial dark matter” that remained largely invisible to classical microbiologists (Schoinas et al., 2023; Wydro, 2022). The emergence of high-throughput sequencing (HTS) and next-generation sequencing (NGS) has dramatically transformed this landscape, offering unprecedented insight into microbial diversity, functional potential, and ecological interactions (Aragona et al., 2022; Huang et al., 2023).
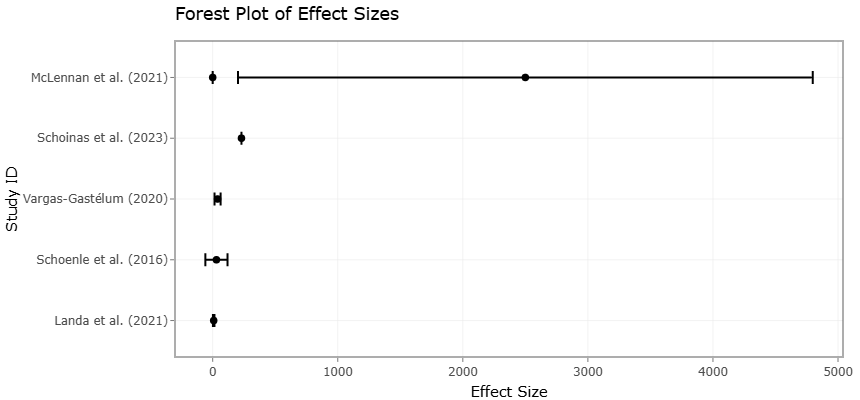
A particularly important application of microbiome research lies in identifying pathogens within environmental reservoirs. In marine systems, the Mediterranean mussel (Mytilus galloprovincialis) exemplifies an effective bioindicator due to its filter-feeding habits. By concentrating environmental particles, including nutrients, heavy metals, and microplastics, these organisms provide a lens through which ecosystem health and pollution levels can be assessed (Figueras et al., 2019; Li et al., 2020). The mussel microbiome is a dynamic consortium that contributes to host nutrition and immunity while simultaneously serving as a habitat for opportunistic pathogens—a phenomenon described as the “pathobiome”. Similarly, extreme environments such as deep-sea sediments and ephemeral rain-fed rock basins in mountains serve as genetic reservoirs for diverse microbial communities, including pathogenic eukaryotes and viruses. These communities have co-evolved under selective pressures unique to their environments, often yielding taxa with unusual metabolic capabilities and virulence strategies (Vargas-Gastélum & Riquelme, 2020; Velasco-González et al., 2023).
The evolution of sequencing technologies has been central to modern pathogen discovery and ecological profiling. Traditional second-generation sequencing (SGS) techniques provide high read depth but are limited in taxonomic resolution due to short read lengths (<550 bp). Third-generation sequencing (TGS) platforms, such as Oxford Nanopore’s MinION and PacBio’s SMRT, offer long-read data that can encompass entire 16S rRNA genes or eukaryotic rDNA operons, enabling species- and even strain-level resolution (Maestri et al., 2019; Winand et al., 2020). These advancements are particularly relevant for pathogens of quarantine concern, allowing for real-time, field-based surveillance of emerging and previously uncharacterized microorganisms (Landa et al., 2021; Huang et al., 2023).
High-fidelity sampling remains a cornerstone of accurate microbiome analysis. Traditional retrieval devices, such as Niskin bottles or piston corers, can alter microbial cell structures and gene expression due to pressure and temperature fluctuations during transit from extreme depths (Edgcomb et al., 2016; Huang et al., 2023). To overcome these challenges, modern sampling approaches employ pressure-retaining and thermally insulated samplers that preserve microbial communities in situ. Instruments like the Microbial Sampler Submersible Incubation Device (MS-SID) and in situ microbial filtration and fixation apparatuses enable immediate stabilization of nucleic acids, allowing downstream analyses to reflect the actual environmental state rather than post-sampling artifacts (He et al., 2023; Huang et al., 2023). Such precision is critical in meta-analyses seeking to quantify pathogen abundance and activity across diverse habitats.
Quantifying microbial pathogens has similarly evolved from culture-based plate counts to highly sensitive molecular techniques. Quantitative PCR (qPCR) remains a standard for monitoring harmful algal blooms and specific fungal pathogens, yet it is prone to interference from environmental inhibitors such as humic acids in soil or sediment matrices (McLennan et al., 2021; Wydro, 2022). Droplet digital PCR (ddPCR) addresses many of these limitations, providing absolute quantification without the need for a standard curve and demonstrating higher tolerance to inhibitors (Kim et al., 2014; Wydro, 2022). Complementary techniques like Catalyzed Reporter Deposition Fluorescence In Situ Hybridization (CARD-FISH) allow for in situ visualization and enumeration of microbial populations, including functional gene expression, enhancing ecological interpretation of pathogen dynamics (Huang et al., 2023; Schoenle et al., 2016).
In the broader framework of microbial community analysis, two major approaches dominate: metabarcoding and shotgun metagenomics. Metabarcoding targets specific taxonomic markers, such as the 16S rRNA gene for bacteria, 18S rRNA (V4 region) for protists, and ITS regions for fungi, providing cost-effective and scalable community profiling (Landa et al., 2021; Nilsson et al., 2019; Vargas-Gastélum & Riquelme, 2020; Velasco-González et al., 2023). However, primer bias and gene copy number variation can limit accuracy. In contrast, shotgun metagenomics sequences total genomic content, offering finer taxonomic resolution down to the strain level and enabling prediction of functional capabilities (Aragona et al., 2022; Knight et al., 2018; Quince et al., 2017). This distinction is particularly important in systematic reviews and meta-analyses that seek to synthesize data across multiple studies with diverse methodological approaches.
Beyond taxonomy, understanding pathogen activity necessitates integrated meta-omics analyses. Metatranscriptomics identifies actively expressed genes, distinguishing living microorganisms from extracellular DNA and providing temporal insight into microbial function (Page & Lawley, 2022; Schoenle et al., 2016). Proteomics and metabolomics, through mass spectrometry and nuclear magnetic resonance (NMR) techniques, reveal the biochemical outputs of microbial interactions, mapping functional landscapes of microbial communities in situ (Huang et al., 2023; Sam et al., 2017). The integration of these datasets, managed through sophisticated bioinformatics pipelines such as QIIME2, DADA2, and CoMA, allows researchers to move beyond mere presence-absence data toward predictive ecological modeling (Aragona et al., 2022; Hupfauf et al., 2020). Importantly, Amplicon Sequence Variants (ASVs) are increasingly preferred over traditional Operational Taxonomic Units (OTUs), providing finer resolution critical for detecting low-abundance pathogens (Callahan et al., 2016; Wydro, 2022).
Analogously, investigating microbial communities without modern HTS and meta-omics tools is akin to attempting to understand the complex social structure of a city by only interviewing a few passersby. Metabarcoding acts as a high-altitude aerial survey, providing a broad overview of residents and their functions, while shotgun metagenomics is equivalent to reading the resumes of every citizen, revealing specialized skills and potential threats. Third-generation sequencing provides a high-resolution lens to distinguish near-identical individuals, and meta transcriptomics functions as a thermal camera, highlighting who is actively engaged in metabolic processes at any given moment. Such technological sophistication allows systematic reviews and meta-analyses to integrate heterogeneous datasets, offering a coherent picture of microbial diversity, pathogen prevalence, and ecological interactions across ecosystems.
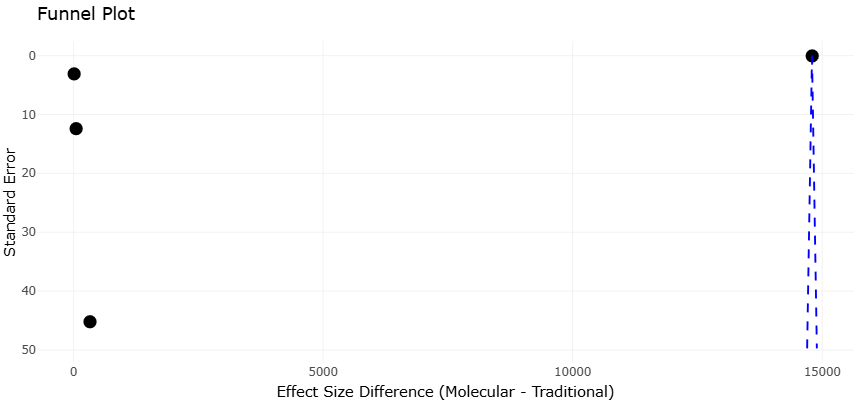
In conclusion, the analysis of microbiomes and associated pathogens has undergone a profound transformation. The integration of high-fidelity sampling, sensitive molecular quantification, advanced sequencing technologies, and meta-omics analyses has shifted the field from a descriptive to a predictive science. Through these approaches, scientists can now access the previously invisible majority of microbial life, identify pathogenic threats in diverse environments, and understand functional dynamics at unprecedented resolution. Systematic review and meta-analysis of these studies underscore the robustness of modern methodologies while highlighting areas for future improvement, including standardization of sampling protocols, mitigation of primer bias, and expansion of reference databases. Collectively, these advancements empower the scientific community to not only map microbial diversity but also anticipate pathogen emergence and ecological impact, fostering a new era in microbiome research.