1. Introduction
Microorganisms form the invisible biological infrastructure of all environment humans inhabit, modify, or exploit. For decades, however, our understanding of microbial ecology was constrained by methodological limitations that favored only those organisms capable of growing under laboratory conditions. This limitation, famously described as the “great plate count anomaly,” suggested that less than 1% of environmental microbes could be cultivated using standard techniques, leaving the vast majority of microbial life uncharacterized (Staley & Konopka, 1985; Pace, 1997). The advent of molecular biology, particularly next-generation sequencing (NGS) and meta-omics approaches, has fundamentally transformed this landscape. These tools now allow researchers to directly interrogate microbial DNA, RNA, and proteins from environmental samples, revealing a staggering diversity of previously hidden taxa and functions across built, clinical, agricultural, and industrial systems.
This paradigm shift has been especially impactful in the study of indoor and engineered environments, where microbial communities are shaped not only by natural ecological processes but also by intense human activity and technological intervention. Built environments such as fitness centers, hospitals, food processing facilities, and wastewater systems function as microbial convergence zones, continuously seeded by human-associated microbiota and subjected to selective pressures such as cleaning regimes, ventilation design, and antimicrobial exposure (Mukherjee et al., 2014; Kembel et al., 2012; Sorgen et al., 2021). These environments are not microbiologically inert; rather, they host dynamic ecosystems whose composition and stability have direct implications for public health, infection control, and the dissemination of antibiotic resistance genes (ARGs).
Fitness centers represent a particularly illustrative example of human-shaped microbial ecosystems. Characterized by frequent skin contact, high occupant turnover, and shared equipment, these spaces accumulate microbial assemblages dominated by human skin–associated taxa, particularly members of the phyla Firmicutes, Proteobacteria, and Actinobacteria (Mukherjee et al., 2014). The dominance of genera such as Staphylococcus, Bacillus, and Pseudomonas reflects continuous microbial deposition from users’ skin, respiratory secretions, and clothing. Similar patterns have been observed on public restroom surfaces and other high-touch indoor environments, underscoring the central role of humans as microbial dispersal agents within built spaces (Flores et al., 2011). Importantly, while many of these microbes are benign commensals, potentially pathogenic species—including Staphylococcus aureus and Klebsiella pneumoniae—can persist on surfaces for extended periods, raising concerns about indirect transmission routes.
Hospitals amplify these concerns due to the vulnerability of their occupants and the concentration of aerosol-generating activities. Healthcare aerosols, defined as suspensions of biological particles in air, are now recognized as critical vectors for nosocomial infections (Matys et al., 2024). Culture-independent studies have revealed that hospital air harbors complex bacterial and fungal communities, including Staphylococcus, Bacillus, Micrococcus, Aspergillus, and Penicillium species, many of which are underestimated or entirely missed by classical culturing methods (Matys et al., 2024). Environmental factors such as ventilation type, seasonality, and human movement strongly influence aerobiome composition, while specific medical procedures—such as dental scaling or patient bathing—generate distinct microbial signatures (Aliabadi et al., 2011; Matys et al., 2024). The integration of molecular surveillance into healthcare settings has therefore become essential for identifying reservoirs of opportunistic pathogens and tracking antibiotic resistance determinants in real time.
Beyond urban indoor spaces, microbial ecology also plays a defining role in industrial and naturalized engineered systems. Wastewater treatment plants (WWTPs), for example, are increasingly viewed as hotspots for microbial evolution and ARG dissemination due to the chronic exposure of microbial communities to sub-inhibitory concentrations of antibiotics and other pharmaceuticals (Lambirth et al., 2018; Sorgen et al., 2021). Similarly, petroleum reservoirs—once thought to be biologically inert—are now recognized as highly active subsurface ecosystems. These environments function as “natural bioreactors,” hosting metabolically diverse microbial consortia capable of hydrocarbon degradation, methanogenesis, and sulfate reduction under extreme conditions of pressure, temperature, and anoxia (Mu & Nazina, 2022; Hidalgo et al., 2021). Global meta-analyses have demonstrated that oil reservoir microbiomes are dominated by Proteobacteria, Firmicutes, and Halobacterota, with a conserved functional core related to energy metabolism and cellular maintenance, despite substantial geographic separation (Hidalgo et al., 2021; Gomes et al., 2023).
Human intervention strongly reshapes these deep subsurface communities. Practices such as water flooding and seawater injection introduce new nutrients and electron acceptors, favoring fast-growing opportunists and sulfate-reducing microorganisms that contribute to reservoir souring and microbiologically influenced corrosion (Mu & Nazina, 2022). These processes mirror ecological dynamics observed in surface-associated biofilms in food processing facilities, where persistent core microbiomes resist sanitation and recolonize equipment surfaces (Xu et al., 2023). Together, these findings highlight that microbial ecology follows consistent principles across vastly different environments, from gym equipment to oil pipelines.
Microbial interactions are equally critical in plant-associated and food systems. Plants host complex microbiomes within their rhizosphere, phyllosphere, and endosphere, where microbial assemblages influence nutrient acquisition, stress tolerance, and disease resistance (Bulgarelli et al., 2013). Studies of olive tree xylem communities demonstrate how plant age, genotype, and pathogen invasion restructure internal microbial networks (Anguita-Maeso et al., 2023). In food systems, culture-independent analyses of fermented foods and sourdoughs reveal food-specific dominance of lactic acid bacteria and yeasts, particularly Lactobacillus and Saccharomyces, which are essential for product quality and safety (De Vuyst et al., 2014; Deka et al., 2021; Syrokou et al., 2020). At the same time, raw vegetables and processing environments act as reservoirs for both beneficial and spoilage-associated microbes, highlighting the thin line between functional fermentation ecosystems and contamination-prone systems (Patz et al., 2019; Xu et al., 2023).
A unifying theme across all these environments is ecological resilience—the balance between microbial stability and adaptability. While certain bacterial communities, such as those on human skin, exhibit long-term stability, other components like the virome display high temporal variability, responding rapidly to environmental change (Hannigan et al., 2015; Hayes et al., 2017). Viruses and bacteriophages play a crucial yet often overlooked role in regulating microbial population dynamics, horizontal gene transfer, and ecosystem function across marine, terrestrial, and built environments (Suttle, 2007; Hayes et al., 2017).
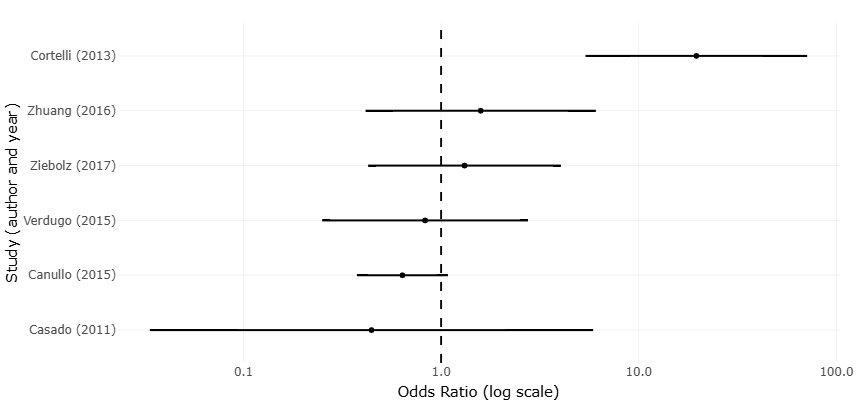
Within clinical microbiology, these advances have profound implications for understanding disease-associated dysbiosis. Peri-implantitis, a biofilm-driven inflammatory disease affecting dental implants, exemplifies the need for molecular approaches to identify consistent microbial biomarkers. Systematic reviews and meta-analyses have demonstrated that PCR-based detection reveals higher prevalence and stronger disease associations for pathogens such as Porphyromonas gingivalis compared to Aggregatibacter actinomycetemcomitans, underscoring the limitations of culture-dependent diagnostics (Belibasakis et al., 2015; Sahrmann et al., 2020). These findings align with broader trends in microbial ecology, where disease risk is increasingly understood as a function of community structure and function rather than the presence of single pathogens.
Despite these advances, significant challenges remain in translating microbial ecological insights into applied solutions. The transfer of plant growth–promoting microorganisms from laboratory discovery to agricultural deployment, for example, continues to face regulatory, ecological, and scalability barriers (Muñoz-Carvajal et al., 2023). Similarly, integrating meta-omics data into routine monitoring of industrial and healthcare environments requires standardized methodologies and interpretive frameworks (Wilmes & Bond, 2004; Hashemi et al., 2021).
In this context, systematic reviews and meta-analyses play a critical role by synthesizing fragmented evidence across disciplines and environments. By integrating data from built, clinical, plant, food, and industrial systems, such approaches provide a holistic view of microbial ecology as a continuum shaped by human behavior, technological intervention, and environmental constraints. This introduction frames the foundation for a systematic review and meta-analysis that examines microbial diversity, transmission pathways, and functional implications across these interconnected ecosystems, highlighting both shared ecological principles and environment-specific risks.