1. Introduction
Microorganisms have long served as nature’s most prolific chemists. For nearly a century, microbial natural products (NPs) have underpinned modern medicine, providing the majority of clinically used antibiotics, antifungals, immunosuppressants, and anticancer agents. The discovery of penicillin from Penicillium by Alexander Fleming marked a turning point in medical history and inaugurated what is often described as the “golden age” of antibiotics (Berdy, 2012; Wright, 2014). In the decades that followed, intensive industrial screening of soil-dwelling microbes—particularly actinomycetes such as Streptomyces—yielded an extraordinary diversity of secondary metabolites, many of which remain cornerstones of contemporary therapeutics (Landwehr et al., 2016; Fischbach & Walsh, 2009). These successes firmly established microbial NPs as a privileged chemical space shaped by evolutionary pressure to interact with biological targets.
Despite this legacy, the effectiveness of traditional discovery pipelines has steadily eroded. The global rise of antimicrobial resistance (AMR) now represents one of the most pressing health challenges of the 21st century. Recent systematic analyses estimate that bacterial AMR was directly responsible for millions of deaths worldwide, with projections suggesting mortality could reach catastrophic levels if new therapies are not developed (Murray et al., 2022). Resistance is driven not only by target modification and efflux mechanisms but also by deeper metabolic rewiring that blunts antibiotic efficacy (Stokes et al., 2019; Lopatkin et al., 2021). At the same time, the antibiotic discovery pipeline has run perilously dry. Classical bioactivity-guided screening approaches increasingly rediscover known molecules rather than uncovering truly novel scaffolds, leading to diminishing returns on investment (Silver, 2008, 2011; Brown & Wright, 2016).
A central reason for this stagnation lies in the mismatch between microbial biosynthetic potential and what is observed under laboratory conditions. Genome sequencing has revealed that most microorganisms encode far more biosynthetic gene clusters (BGCs) than the number of metabolites they produce in culture. It is now widely accepted that approximately 90% of microbial BGCs are transcriptionally silent or poorly expressed under standard cultivation conditions, effectively concealing a vast reservoir of “biosynthetic dark matter” (Rutledge & Challis, 2015; Baltz, 2017). Traditional top-down discovery strategies—cultivate, extract, screen—are inherently blind to this hidden potential, as they depend on the fortuitous expression of pathways that microbes may only activate in specific ecological contexts.
In response to these limitations, microbial NP discovery has undergone a profound conceptual shift. Rather than beginning with molecules, modern strategies increasingly start with genes. Genomics-driven, or “bottom-up,” discovery leverages high-throughput sequencing to systematically explore microbial genomes for BGCs, the physical groupings of genes responsible for secondary metabolite biosynthesis (Ziemert et al., 2016; Albarano et al., 2020). This shift has transformed discovery from a largely serendipitous endeavor into a data-driven science, enabling rational prioritization of strains, pathways, and environments with the highest likelihood of yielding novel chemistry (Malit et al., 2022; Rosic, 2022).
Bioinformatic platforms such as antiSMASH have become foundational tools in this new paradigm, providing automated detection and annotation of BGCs across bacterial and fungal genomes (Ziemert et al., 2016; Meena et al., 2024). Complementary approaches now integrate resistance-gene–based logic, recognizing that producers of bioactive compounds must encode self-resistance mechanisms to avoid autotoxicity. Databases and tools such as ARTS and CARD exploit this principle, allowing researchers to infer potential modes of action directly from genomic data and thereby prioritize BGCs with therapeutic relevance (Dong & Ming, 2023; Alcock et al., 2023; Mungan et al., 2022; O’Neill et al., 2019). From a systematic review perspective, these strategies collectively represent a shift from random screening toward hypothesis-driven mining grounded in evolutionary and functional logic.
Equally transformative has been the expansion of discovery beyond cultivable microbes. Environmental sequencing has revealed extraordinary microbial diversity, much of which resides in the so-called “rare biosphere” and remains inaccessible to classical microbiology (Sogin et al., 2006). Metagenomics circumvents this barrier by extracting and sequencing environmental DNA directly from complex habitats such as soils, sediments, and marine organisms (Handelsman, 2004; Venter et al., 2004). This approach has dramatically expanded the searchable biosynthetic landscape and enabled the reconstruction of BGCs from entire microbial communities rather than isolated strains (Rosic, 2022). Notably, culture-independent and in situ cultivation technologies, such as the isolation chip (iChip), have bridged the gap between genomics and chemistry, culminating in landmark discoveries such as teixobactin—a compound with no detectable resistance to date (Ling et al., 2015).
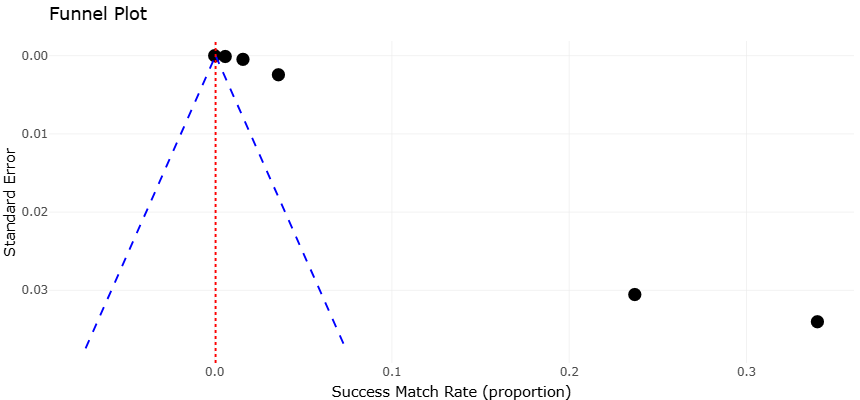
The marine environment has emerged as a particularly rich frontier for genome mining and metagenomics-based discovery. Marine microbes experience unique ecological pressures that drive the evolution of chemically distinct metabolites, many of which display potent antibacterial or anticancer activities (Wietz et al., 2010; Albarano et al., 2020). Systematic surveys of marine-derived genomes have revealed thousands of previously uncharacterized gene cluster families, underscoring how little of this biosynthetic space has been explored (Rosic, 2022). From a meta-analytical viewpoint, marine BGCs show lower match rates to known compounds compared with terrestrial counterparts, reinforcing their value as sources of chemical novelty.
The current “deep mining” era extends beyond genomics alone by integrating metabolomics, proteomics, and computational analytics into unified discovery workflows (Gaudêncio et al., 2023; Wang et al., 2025). Mass spectrometry–based molecular networking platforms such as GNPS allow researchers to rapidly dereplicate known metabolites and visualize chemical relatedness across large datasets, reducing redundancy and bias in compound selection (Wang et al., 2016). These tools are particularly powerful when combined with genome mining, as they help establish gene–metabolite links that are essential for validating BGC predictions.
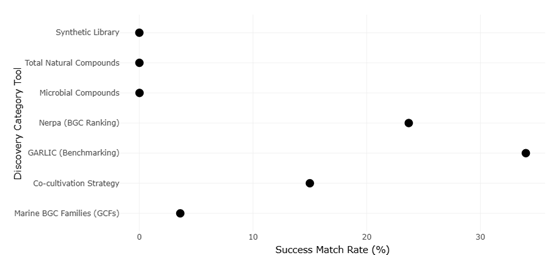
Artificial intelligence (AI) and machine learning (ML) now play a central role in managing the scale and complexity of modern discovery data. Deep learning models have improved the detection, classification, and prioritization of BGCs, outperforming traditional rule-based algorithms (Gaudêncio et al., 2023; Wang et al., 2025). Specialized tools such as Nerpa further exemplify this trend by enabling high-throughput matching of known nonribosomal peptide structures against thousands of genomes, thereby accelerating the identification of novel or divergent biosynthetic pathways (Kunyavskaya et al., 2021). Collectively, these advances are reshaping NP discovery into a predictive science that connects genotype, chemotype, and phenotype with unprecedented resolution.
Viewed through the lens of systematic review and meta-analysis, the accumulated evidence clearly indicates that genomics-driven and integrative discovery strategies outperform traditional approaches in terms of novelty yield and mechanistic insight. While challenges remain—particularly in translating genomic predictions into scalable compound production—the convergence of genome mining, metagenomics, high-throughput analytics, and AI offers a credible path forward. In an era defined by escalating antimicrobial resistance, revisiting microbial natural products through deep mining is not merely an academic exercise but a necessity for sustaining the future of anti-infective drug discovery.